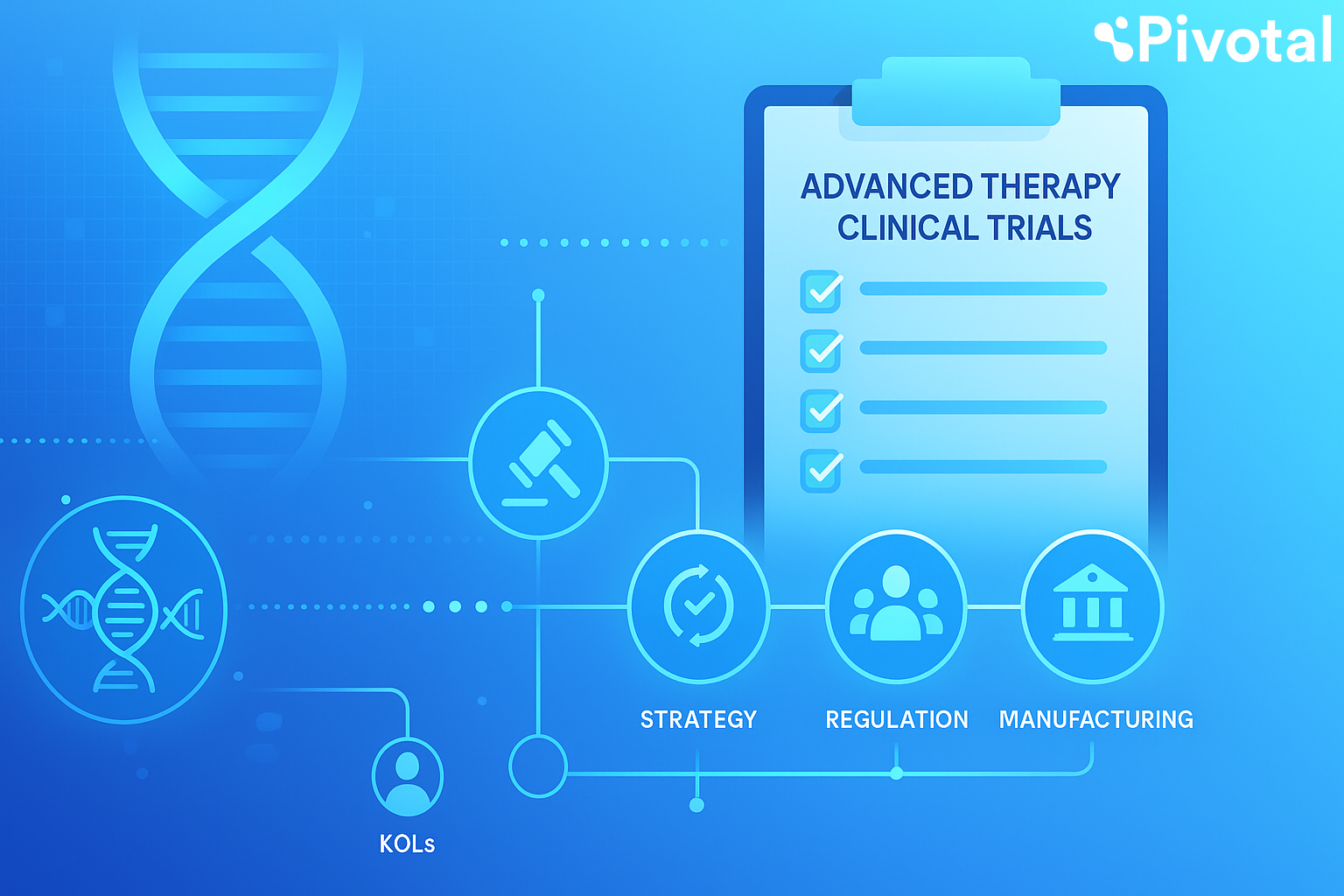
Advanced therapy medicinal products (ATMPs)—including gene therapy medicinal products (GTMPs), somatic cell therapy medicinal products (sCTMPs), tissue-engineered products (TEPs), and combined ATMPs (e.g. cells or tissue with a device)—are transforming modern medicine. These therapies present unprecedented opportunities, particularly for rare, orphan, or complex diseases.
However, the path to clinical development is challenging, requiring precise planning, deep expertise, and the right partners. This checklist outlines the critical steps and timelines for early-phase (Phase 1 and 2) ATMP trials, with a particular focus on EU and UK regulatory frameworks.
1. Pre-Trial Planning (6-12 months before trial start)
- Collaborate with Key Opinion Leaders (KOLs)
Engage experienced KOLs early to shape your trial design, endpoints, and patient selection criteria. Their expertise is essential for addressing the scientific, regulatory, and ethical nuances of ATMPs.
- Involve Patient Advocacy Groups (PAGs)
Especially for rare diseases, working with advocacy groups helps ensure your trial is patient-centric. Their input on aspects such as the schedule of assessments can improve protocol feasibility and a patient engagement.
- Regulatory Roadmap for ATMPs
When planning your trial, key actions include:
-
- Request Scientific Advice from EMA or MHRA to align expectations on nonclinical and clinical data.
- Classify Your Product as an ATMP by submitting a formal ATMP classification request (EMA or MHRA). While this is optional and non-binding, it can unlock access to incentives.
- Preparing for a Clinical Trial Application (CTA). Unlike the IND process in the US, the EU and UK require a CTA, which includes the Investigational Medicinal Product Dossier (IMPD) (see Section 4.).
- Assess the Competitive Landscape
Study the pipeline for comparable ATMPs, focusing on their safety, efficacy, target population, and trial designs. Use the data to position your therapy effectively and anticipate regulatory or clinical hurdles.
When: Begin 12 months prior to FPI and review regularly.
2. Manufacturing and Logistics (6-12 months before trial start)
- Confirm Intellectual Property (IP) & Manufacturing Strategy
Protect critical elements such as vectors, cell lines, or gene editing tools. Choose a GMP-certified manufacturer with ATMP experience.
Tip: Keep the end goal in mind. Even if batch sizes in early trials are small, consider how IMP manufacturing could be scaled for late-stage trials and commercialization.
When: Finalize at least 9-12 months before trial.
3. Vendor and CRO Selection (6-9 months before trial start)
- Choose Experienced Vendors
Prioritize partners with a strong track record in:
-
- Biologics and cell-based therapies.
- Specialized assay development (PK, immunogenicity).
- Cold-chain and cross-border logistics, including EU-specific customs processes and UK-specific regulations.
- Partner with a Specialized Clinical CRO
ATMPs demand a CRO with deep expertise, robust quality systems, and established relationships with regulatory agencies. Evaluate CROs based on their history with similar therapies, operational flexibility, and access to the latest technology platforms (e.g. decentralized trial solutions) as well as their accessibility and responsiveness.
This is where a trusted partner like Pivotal makes a difference. We offer:
-
- Medical, Operational and Regulatory expertise in early phase trials and ATMPs.
- Decentralized trial capabilities, including remote monitoring and digital patient engagement tools.
- Patient Recruitment and Retention who collaborate with advocacy groups and KOLs to meet enrolment milestones.
- Prequalified ‘golden sites’, with proven experience in ATMP trials and high-quality data capture.
When: Begin evaluating vendors 9 months before trial start off and finalize by 6 months before trial start off.
4. Protocol Development and Trial Setup (4-6 months before trial start)
- Prepare the CTA and IMPD for Advanced Therapies
The CTA must be submitted to regulatory authorities (e.g. EMA, MHRA) and ethics committees. The submission will include the IMPD, which outlines:
-
- Product composition and manufacturing process.
- Preclinical safety and efficacy data.
- Proposed trial design and safety monitoring plan.
Tip: For ATMPs, include a detailed Risk-Based Approach, in line with EMA guidance, addressing product-specific risks such as immunogenicity and off-target effects. Long-term follow-up (LTFU) is also essential due to durable therapeutic effects – build this upfront into your trial design and within your regulatory strategy.
Tip: Tailor the Protocol Design to ATMPs. Incorporate adaptive designs and innovative endpoints (e.g. biomarkers). Input from KOLs, advocacy groups, and your trusted CRO ensures the design is both regulatory-compliant and patient-friendly.
When: Begin drafting 6 months in advance.
5. Pre-Trial Execution (1-3 months before trial start)
- Secure GMO Clearance (If Applicable)
Gene therapies often involve genetically modified organisms (GMOs), which require environmental and safety assessments under EU and UK GMO regulations. Submit applications early to avoid delays.
When: Start clearance process 3-6 months before trial start off.
- Ensure Site Readiness
Train investigators and site staff on handling, storing, and administering advanced therapies, as well as monitoring for adverse events such as cytokine release syndrome. Sites should meet GMP handling standards.
- Validate Data Capture and Monitoring Systems
Implement electronic data capture (EDC) and monitoring systems that comply with EU’s Annex 11 and UK data integrity requirements.
When: Begin training and system validation 3 months prior to FPI.
6. Ongoing Trial Management (During and Post-Trial)
- Monitor Safety with Real-Time Data
Real-time monitoring is vital due to ATMPs’ complex safety profiles. Use platforms such as Pivotal’s Danah for immediate insights and early detection of adverse events.
- Maintain Long-Term Traceability
EU regulations require traceability of ATMPs from donation to administration for 30 years. In the UK, similar long-term tracking requirements apply. Implement robust systems to manage this requirement.
- Submit Periodic Safety Updates (PSURs)
Regular safety reporting is required by both EMA and MHRA for ongoing trials, particularly for therapies with unique safety profiles. Establish systems to streamline this process.
Summary and Next Steps:
Early phase advanced therapy trials require meticulous planning across regulatory, operational, and scientific domains. By following this tailored checklist, drug developers will navigate the path more confidently – and increase the likelihood of trial success.
Here at Pivotal we specialize in supporting Biotech Companies throughout the life span of trial preparation and execution and during every stage of ATMP clinical development.
Why partner with Pivotal for your ATMP trial?
- Focused on Biotech: we understand the unique challenges small teams face, and offer flexible, responsive support tailored to your needs.
- Scientific Depth: our team includes 12 MDs with hands-on experience in ATMP trial strategy and execution.
- End-to-end patient engagement: our operations and patient recruitment and retention teams will leverage decentralized solutions, KOL networks, and PAGs to drive robust recruitment metrics.
If you’re currently planning your trial and would like to talk to our experts, reach out to our team.
MEDIA CONTACT
Ms. Natalia Farr